
猜你喜欢
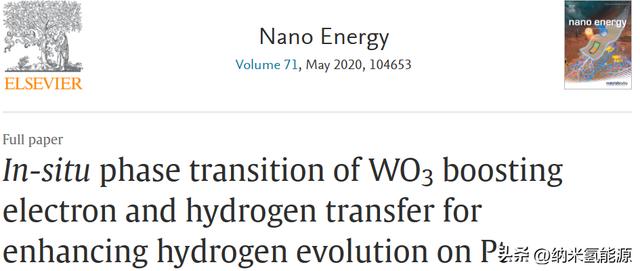
图1 Pt-WO3和WO3的XRD图谱。Pt-WO3的Pt 4f XPS光谱。Pt-WO3的O 1s XPS光谱。Pt-WO3的暗场STEM图像。具有清晰格子条纹的相应STEM图像。快速傅里叶变换滤波后Pt-WO3的HAADF图像。
首先,采用共沉淀法合成氧化钨纳米片,然后在大气下煅烧;随后,在室温水溶液中通过光化学方法将Pt沉积在氧化钨纳米片(Pt-WO3)上。这种简单温和的光化学方法能够控制Pt在WO3上的负载,合成各种配比的Pt-WO3催化剂。Pt-WO3的X射线衍射(XRD)图谱与WO3的X射线衍射(XRD)图样(PDF#83–0951)相匹配,没有可辨别的铂衍射峰,表明Pt-WO3中不存在Pt的晶体纳米结构。
采用X射线光电子能谱(XPS)研究了Pt-WO3的价态和化学键信息。Pt-WO3中的Pt原子主要以 2和 4形式存在。结合能为78.1 eV的峰可以分配给Pt4 的4f7/2,72.6 eV和76.0 eV处的两个峰分别属于Pt2 的4f5/2和4f7/2。有趣的是,金属Pt0的缺失表明催化剂中没有Pt纳米颗粒。此外,图中O 1s XPS光谱中的两个峰。1c可以归因于W-O键(530.3 eV)和Pt-O键(530.9 eV),这进一步验证了Pt原子以阳离子形式存在,而不是金属状态。Pt-O键的形成可归因于光生空穴氧化了PtCl62−中的Cl−,而光生电子将Pt4 还原为Pt2 。这些结果表明, 铂原子或团簇通过与表面氧原子键合加载到WO3上。
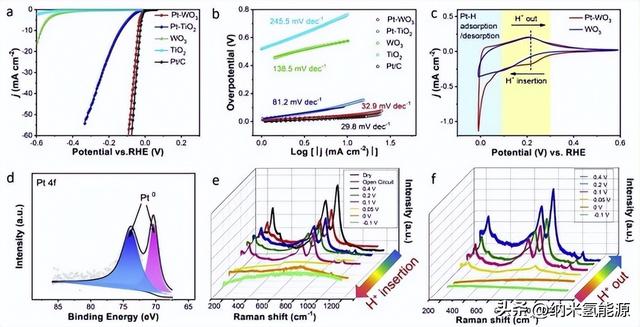
图2 Pt-WO3的LSV曲线和HER在0.5 M H 2 SO4溶液中的比较催化剂。(b) 催化剂的塔菲尔斜率。Pt-WO3和WO3的CV曲线在0.59和-0.01V之间(相对于RHE)。(d)Pt-WO3在5000次CV循环后Pt 4f的XPS光谱。(e-f) Pt-WO3 的原位拉曼光谱从 0.4 到 −0.1 V in (e) 并扫描回 (f)。
在0.5 M H2SO4水溶液中考察了一系列催化剂的HER性能。如图2a所示,Pt-WO3表现出优异的HER性能,接近商业20 wt% Pt/C催化剂的性能。在极化电流密度为10 mA cm−2时,Pt-WO3的过电位达到39 mV,仅比Pt/C的28 mV高11 mV。
为进一步研究Pt-WO3在电催化条件下的催化活性位点和结构演变,采用循环伏安法(CV)表征了催化剂在0.5 M H2SO4溶液中的电化学行为。 如图所示。如图2c所示,Pt-WO3和WO3的CV曲线表现出不同的氧化还原峰。 根据以前的工作,WO3的氧化还原峰在0.3和0 V之间可以归因于WO3中的H 插入和插入。 当H 以电子插入WO3时,WO3逐渐转变为非化学计量氧化物:HxWO3,即钨青铜。 另一方面,Pt-WO3在0.3至0.1 V之间的电位区域的明显电流增加主要是由于Pt上特定的氢吸附。同时,Pt负载Pt-WO3在0.2V左右的还原峰正偏移使H 插入/拔出过程更具可逆性。 在较低电位(0.1V至−0.01 V)下,氢吸附中间体的形成主要导致电流增加,这证明了Pt-WO3中的Pt原子是HER的活性位点。 作为比较,图中的CV曲线。S8表现出Pt–TiO2的低氢吸附电流,表明Pt/TiO2的氢吸附能力较差,H 插入效应是WO3的独特效应。 因此,金属氧化物载体的性质将极大地影响催化剂的电化学氢吸附行为,从而极大地决定其HER活性。此外,通过XPS测量表征了HER循环后Pt-WO3的表面化学状态。在图2d中,Pt 4f XPS谱显示Pt0组分是催化HER时真正的活性位点。催化还原前Pt价由 2或 4原位还原为0的原因是Pt 2 /Pt和Pt 4 /Pt的还原电位高于H /H2。从图S9中,530.3 eV处的W-O特征峰可以看出,在HER测试前后,WO3的表面结构基本保持不变。同时,O1s谱中530.8 V处的峰可分配给W-O-H键,这可能是由于残留的氢离子被困在WO3中造成的。虽然Pt-WO3中的Pt被还原为Pt0,但经过HER测试后,Pt-WO3的XRD图中没有明显的晶体Pt衍射峰(图S10)。此外,在图S11中,经过HER后的Pt-WO3的HRTEM图像显示了良好的WO3晶体结构,没有发现明显的Pt纳米颗粒。可以推断出Pt-WO3的真正活性位点是小的Pt团簇,没有形成块体结构。
为了进一步验证WO3在电化学循环过程中H 的插入和相变,进行了原位拉曼光谱测量。如图2e和f所示,在268 cm−1,710 cm−1和806 cm−1处的峰可以被划分为WO3表面W-O键的特征峰。WO3的拉曼特征峰随着施加的电位从0.1 V到0.05 V(相对于RHE)消失,然后随着电位向后扫而重新出现。这一观察结果验证了Pt-WO3的可逆相变,这与图2c CV曲线所示的H 在WO3中的插入和退出相吻合。此外,Pt-WO3催化剂的颜色随着电位负移逐渐由黄色转变为蓝色,当电位恢复到开始时颜色又恢复。根据上述结果,原位电化学还原金属Pt团簇锚定在WO3上被证明是Pt-WO3催化HER的真正活性位点,并验证了电化学H 插入/输出过程在WO3与HxWO3之间的原位相变。
性能测试
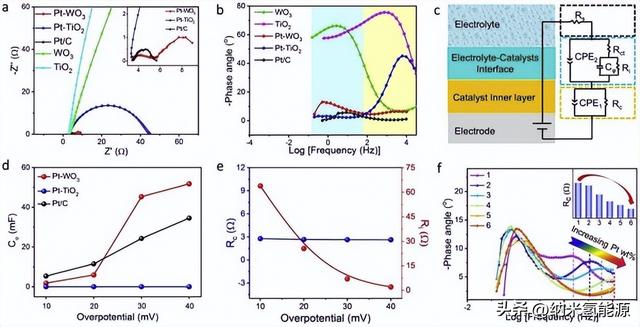
图3 Pt-WO3和参比催化剂在40 mV过电位下的奈奎斯特图。(a)中催化剂的相应波特图。(c). Pt-WO3电极结构及等效电路示意图.(d). Pt-WO3、Pt–TiO2 和 Pt/C 在不同过电位 (10 mV–40 mV) 下的 Cφ 拟合数据。不同过电位(10 mV–40 mV)下Rc(蓝色)和Ri(红色)的拟合数据。(f) 过电位为 40 mV 的 Pt-WO3 催化剂组的波特图,插图中带有相应的 Rc 拟合值。
为了进一步探究Pt-WO3的催化机理,采用原位电化学阻抗谱(EIS)技术进行了研究。首先,用EIS对一组Pt-WO3和参比催化剂在不同应用反应电位下进行了表征。S12-S21)。如图3a所示,在HER过电位为40 mV时采集Nyquist图。显然,Pt-WO3和Pt/C的圆弧小得多,表明其电极系统中的电阻小得多。奈奎斯特图呈圆弧状而不是完美的半圆形,通常归因于催化剂电极表面原子粗糙和垂直多孔结构(由纳米材料在电极上无序堆积引起)。如图所示,Pt-WO3的Nyquist图包含两条弧段,而其他图只包含一条弧段。两种不同类型的电化学过程可以产生两个特征频率不同的半圆。图3b为对应的波德图,图中显示了相位角随频率变化的趋势。Bode图可分为高频区(黄色)和低频区(青色)两个区域。根据前人的工作,高频区和低频区信号分别为催化剂内层/界面的电子传导和电解质-催化剂界面上的反应电荷转移。结果表明,Pt基催化剂在两个频率区表现出不同的行为。其中,Pt-TiO2的高频区高相角和低频区极低相角说明HER的电子转移电阻主要来自催化剂内层电子转移电阻,而不是电解质-催化剂界面电荷转移电阻。对于Pt-WO3,在高频区相角较低,表明Pt-WO3具有更快的电子转移,界面电荷转移主导了HER的反应电阻。同样,Pt/C催化剂由于金属和碳载体优异的电子导电性,仅受界面电荷转移电阻的限制。此外,WO3和TiO2的对比也证实了WO3中的电子转移比TiO2快得多。由此证明Pt-WO3中快速的内层电子转移促进了HER反应速率,而Hads吸附/解吸的速率限制过程是由Pt活性位点的内在活性决定的。
随后,我们使用等效电路模型(图3c)将溶液电阻(Rs)串联成两段来模拟EIS数据(见表S3)。含有电阻(Rc)和恒相元件(CPE1)的并联电路的第一段对应于催化剂内层电路。第二部分对应于一个反应中间积累过程(Ri和Cϕ)中的电解质-催化剂界面反应电荷转移过程(Rct和CPE2)。从数据上看,Pt-TiO2的电阻以Rc为主,Pt-WO3的电阻主要由Ri贡献。正是如此,这一发现与图3b中不同预兆图类型的结果一致。cφ值表示氢中间体(Hads)吸附伪电容,反映Hads在催化剂活性位点上的覆盖范围。如图3d所示,Pt-WO3的cφ值越高,表明Pt-WO3的Hads在Pt上的覆盖率要比Pt-TiO2 (甚至Pt/C)高得多,这有利于HER动力学。根据前人对HxWO3的研究,该钨青铜相是一种金属模拟非化学计量氧化物,由于W与移动的氢离子容易进行电子转移,具有优异的电子和氢转移能力。这种高Hads覆盖率可归因于电化学原位形成的HxWO3相具有较强的电子转移能力。同样,Pt-WO3的小Rc也验证了HxWO3在反应过程中的加速电子转移效应。此外,图3e总结了Pt-WO3在各种过电位下的拟合Rc和Ri。随着过电位的增加,Ri逐渐减小,而Rc基本保持不变,证明Rc是由催化剂的固有性质决定的。从图3f的Bode图可以看出,随着Pt含量的增加,Pt-WO3催化剂的高频区相位角逐渐减小,并向高频方向移动,说明随着Pt含量的增加,Pt-WO3催化剂中的电子转移速率加快。此外,插图还揭示了Rc值随着Pt含量的增加而降低。这种现象可以归因于铂团簇或纳米颗粒具有优异的电子转移能力。由此可见,随着Pt含量的增加,Pt/HxWO3界面的形成可以进一步提高Pt-WO3催化剂的电子转移能力。
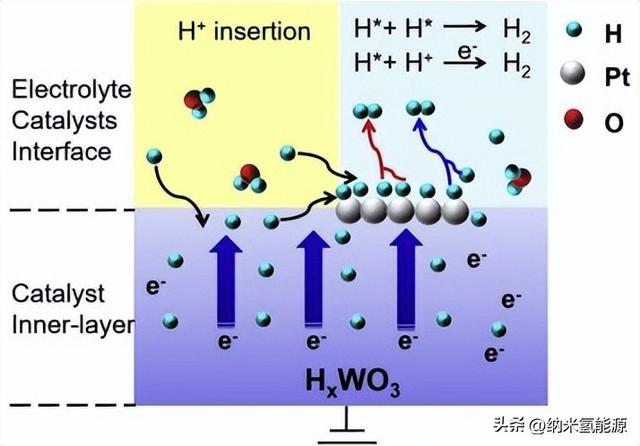
图4 简要说明了Pt/WO3的促进机制。
根据上述结果,对Pt-WO3具有优异的HER活性的原因进行了探讨。如图4所示,H 首先与电子耦合插入WO3中,在负电位作用下形成钨青铜HxWO3。由于类似的金属性质,H 很容易在HxWO3中迁移并伴随电子转移,从而使HxWO3在电化学过程中成为一个快速的电子/氢转移通道,促进电子和氢的转移。此外,HxWO3作为真正的载体,不仅加速了反应中的电子转移,而且可以通过Pt/HxWO3界面打开一个H 转移到催化活性位点的途径。这样可以大大提高催化剂上Hads的覆盖率,从根本上促进HER动力学。因此,HxWO3优异的电子-氢转移能力极大地保证了电子注入到催化剂表面活性位点,促进了氢向Pt的转移,从而具有优异的HER活性。这种对促进氢转移的认识与没有实验证据的氢溢出效应促进HER机制不同。
结论
综上所述,成功合成了一系列在WO3上负载铂簇的催化剂,在低铂含量的酸性溶液中表现出较高的催化活性和稳定性。采用原位拉曼、EIS等一系列光谱分析方法,通过Pt/TiO2和Pt/C的对比,深入研究了WO3对Pt催化氧化的活性位点、结构演化以及WO3对Pt催化氧化的促进作用。HER后催化剂的原位拉曼光谱、CV曲线和XPS谱证实了Pt团簇是真正的活性位点,Pt-WO3中存在电化学驱动的由WO3向HxWO3的相变。催化剂的原位EIS结果表明,HxWO3在Pt-WO3中进行了快速电子转移,促进了HER过程中的电子转移。进一步表征Au和Pd基催化剂的对比表明,HxWO3加速了氢向Pt的转移,提高了反应中间体Hads的覆盖率,从而改善了HER动力学。本文证实了HxWO3对Pt提高HER性能的作用源于HxWO3的快速电子转移以及HxWO3向Pt的氢转移途径。该工作验证了电催化反应中催化剂的结构演化,证明了电子/氢转移在电催化HER中的重要作用。
参考文献
Chao Xie, et al. In-situ Phase Transition of WO3 Boosting Electron and Hydrogen Transfer for Enhancing Hydrogen Evolution on Pt. Nano Energy, 2020.













 智钛公众号
智钛公众号 智钛小程序
智钛小程序