
猜你喜欢
用于稳定原子分散催化剂的功能性CeOx纳米胶
文章出处:Xu Li, Xavier Isidro Pereira-Hernández, Yizhen Chen, Jia Xu, Jiankang Zhao, Chih-Wen Pao, Chia-Yu Fang, Jie Zeng, Yong Wang, Bruce C. Gates, Jingyue Liu. Functional CeOx nanoglues for robust atomically dispersed catalysts. Nature 2022, 611, 284-288.
摘要:单原子催化剂能非常有效地利用昂贵的贵金属,并能产生独特的性能。然而,由于烧结的原因,催化剂的稳定性有限,通常会影响催化剂的应用。尽管将金属原子锚定在氧化物载体上可以抑制烧结,但强的金属-氧相互作用往往留给反应物结合和催化的金属位点太少,当暴露在足够高的还原温度下时,即使是氧化物锚定的单原子催化剂最终也会烧结。在这里,作者展示了通过将原子分散的金属原子限制在氧化物纳米簇或“纳米胶”上可以增强锚定的有益效应,这些纳米胶本身分散并固定在一个坚固的、高表面积的载体上。作者通过将孤立的、有缺陷的CeOx纳米胶岛嫁接到高表面积的SiO2上证明了这一策略;每个纳米胶岛平均拥有一个Pt原子。作者发现,Pt原子在高温氧化和还原环境下都保持分散,活化催化剂对CO的氧化活性显著提高。作者将还原条件下提高的稳定性归因于支撑结构和Pt原子对CeOx比SiO2更强的亲和力,这确保了Pt原子可以移动,但仍被限制在各自的纳米胶岛中。使用功能性纳米胶来限制原子分散的金属,同时增强其反应性的策略是普遍的,作者预计它将使单原子催化剂更接近实际应用。
该设计策略将三种成分整合到催化剂中:(1) 坚固的高表面积载体(例如SiO2,Al2O3),(2) 金属氧化物纳米胶团簇(例如CeOx,TiOx)作为孤岛稳定地固定在载体上,(3) 单金属原子(M1)定位在这些孤岛上。纳米胶的选择标准包括它们在载体上以分散形式的稳定性,它们对活性金属原子的亲和力比载体强得多,以及它们与活性金属的相互作用以增强催化性能。
SiO2是一种不可还原的、廉价的、广泛应用的催化剂载体,具有高表面积、结构稳定和商业价值。由于金属原子通常通过强的M1-Ox键固定在缺陷位点的可还原金属氧化物(例如CeO2、TiO2)上,并且由于CeO2具有宝贵的氧化还原和储氧性能,作者选择CeOx纳米团簇作为原型纳米胶来定位CO氧化的Pt原子。
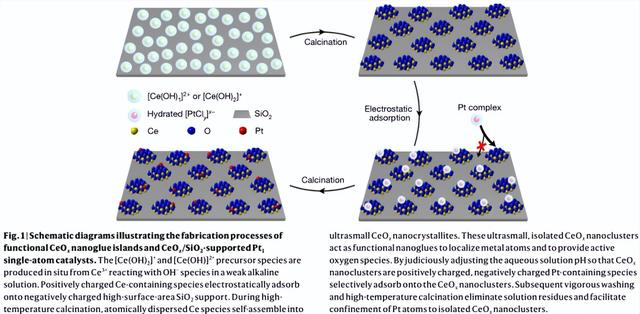
图2
X射线吸收光谱进一步揭示了Pt的性质。Pt LIII边缘X射线吸收近边缘结构(XANES)数据(图3a)表征了焙烧的0.4 wt.% Pt/CeOx/SiO2,其结果表明Pt为阳离子,其氧化态接近于体态PtO2。扩展X射线吸收精细结构(EXAFS)光谱(图3b)表明Pt原子分散,在0.4 wt.% Pt/CeOx/SiO2的光谱中没有Pt-Pt散射路径的证据(图3b)。Pt-O壳层表明Pt-CeOx键合,配位数为4.5 ± 0.5,键合距离为1.97 ± 0.02 Å,与原位分离Pt在CeO2或Fe2O3上的EXAFS数据一致。漫反射红外傅立叶变换光谱(DRIFTS)显示0.4 wt.% Pt/CeOx/SiO2在CO吸附后,在2103 cm-1附近出现了一个尖锐的单峰,半峰值全宽为12.8 cm-1 (图3c),属于吸附在孤立阳离子Pt上的CO,与XANES和EXAFS的结果一致。
为了进一步了解原子分散Pt的稳定性,比较了Pt原子在不同条件下在其它载体上的行为。数据包括在SiO2和CeO2上获得的Pt原子。在高于300 oC的还原或氧化环境下,SiO2上的Pt原子大量烧结,显示出Pt-SiO2之间的弱相互作用。CeO2 NPs上的单个Pt原子在焙烧过程中没有烧结,即使在高温下也是如此。然而,在300 oC下暴露于H2中1小时,导致Pt-O键断裂,Pt原子迁移到CeO2 NPs上,Pt原子烧结形成团簇和NPs。相同处理后,在CeO2上形成的Pt NPs小于在SiO2上形成的Pt NPs,说明在H2还原处理过程中Pt-CeO2相互作用强于Pt-SiO2相互作用。对混杂在CeO2-SiO2核壳模型体系中的Pt的研究证实,Pt原子与CeO2的相互作用强于与SiO2的相互作用,从而抑制了Pt从CeO2向SiO2的扩散。
研究了CeOx负载Pt单原子催化剂(Pt1/CeOx/SiO2)在还原条件下的稳定性。根据DRIFTS和HAADF-STEM数据(图3d),CeO2 NP负载的Pt原子在高于300 oC的温度下在H2中持续1小时可移动,但即使在300 oC的H2还原10小时后,仅局限于孤立的CeOx岛的Pt原子也没有烧结。表征还原催化剂和合成催化剂中Pt原子的CO吸收带的最小差异证明了在DRIFTS实验中使用的CO探针分子还原了合成的Pt1/CeOx/SiO2中的Pt原子,如预期的那样。为了进一步探测CeOx岛上Pt原子的稳定性,样品在400-600 oC的温度下暴露在H2中。即使在这些苛刻的还原条件下,Pt仍然是孤立的单原子。表征Pt原子上CO的红外吸收峰在高于500 oC的温度下被H2还原后发生了偏移,表明Pt-CeOx相互作用发生了改变,这被密度泛函理论计算所证实。由于DRIFTS实验是在无氧环境下进行的,因此排除了PtxOy团簇的形成。Ce XPS和Pt XAS数据均表明,在还原条件下没有形成Pt团簇或Pt-Ce合金物种。因此,尽管催化剂发生了变化,Pt位点仍然是孤立的,变化仅限于载体CeOx岛。相比之下,CeO2载体上Pt原子数密度的降低(其负载量低于0.4 wt.% Pt1/CeOx/SiO2的负载量)不能阻止Pt原子在400 oC的H2还原后形成团簇和NPs
为了证实在H2还原过程中,SiO2负载的CeOx岛上的Pt约束也适用于SiO2负载的CeO2 NPs,一个含有Pt原子分散在SiO2上的样品负载8 nm的CeO2 NPs (0.4 wt.% Pt1/CeOx/SiO2)在300 oC的H2中暴露1小时。数据表明,Pt原子没有迁移到SiO2上,而是烧结形成了小的Pt团簇,因为较大的CeO2 NPs平均包含一个以上的Pt原子,进一步证实了还原条件下Pt原子在CeO2表面烧结的结论。相应的CO的DRIFTS数据显示CO吸附在Pt团簇和NPs上。与以0.4 wt.% Pt1/CeOx/SiO2为表征的狭窄DRIFTS峰(图3d)形成鲜明对比的是,以0.4 wt.% Pt/CeO2 NPs/SiO2为表征的宽阔的重叠DRIFTS峰显示了具有广泛核性和/或氧化态的Pt物种的存在。SiO2对CeOx纳米胶岛的分离和超细尺寸是限制Pt种类以防止烧结的关键。
在SiO2负载的CeOx纳米岛上进行了高负载Pt (4 wt.%)的进一步实验,研究了均匀Pt簇的合成和约束。在300 oC的H2还原3小时后,通过CO DRIFTS检测到Pt团簇。HAADF-STEM图像显示,当还原温度升高到400 oC时,孤立的CeOx岛上附着了均匀的Pt团簇(平均大小约0.9 nm)。在500 oC还原12小时后,CeOx岛变成了无定形,但Pt团簇仍然保持了它们的大小,并仍然附着在岛上。在这些严重还原的高Pt负载催化剂中,尺寸大于1 nm的Pt NPs的缺失突出了CeOx纳米胶策略在稳定定位Pt (从孤立的金属原子到亚纳米簇)和防止形成更大的Pt NPs方面的有效性,从而扩大了其实际应用潜力。
作者注意到,许多催化剂中的活性相是通过还原预先形成的,在高于200 oC的温度下H2处理通常会导致原子分散的贵金属的烧结。除了限制应用之外,这种结构调整也阻碍了单原子催化剂性能和结构-性能关系的基础研究。0.4 wt.% Pt/CeOx/SiO2在氧化和还原环境下的稳定性为这方面提供了新的机会。

图3
据报道,Ce负载的单Pt原子对CO氧化的活性低于CeO2负载的Pt团簇,作者的数据证实了这一模式。由于作者的CeOx纳米岛限制Pt单原子催化剂在还原活化后仍保持稳定,作者可以探索CO氧化性能,并显示H2活化的Pt1/CeOx/SiO2的50% CO转化温度为133 oC (图4),未活化的Pt1/CeOx/SiO2为226 oC。结果表明,H2活化使CO氧化速率提高了两个数量级,并使表观活化能降低。XANES和EXAFS数据表明,H2还原去除了氧配体,使Pt-O第一壳层的配位数从4.5 ± 0.5降低到3.2 ± 0.3,使Pt4 还原为Ptδ ,而无需烧结分离的Pt原子。与在其它CeO2载体上制备的催化剂或用Pt盐溶液浸渍CeOx/SiO2和SiO2制备的催化剂相比,H2活化的0.4 wt.% Pt1/CeOx/SiO2对CO的氧化活性更稳定。
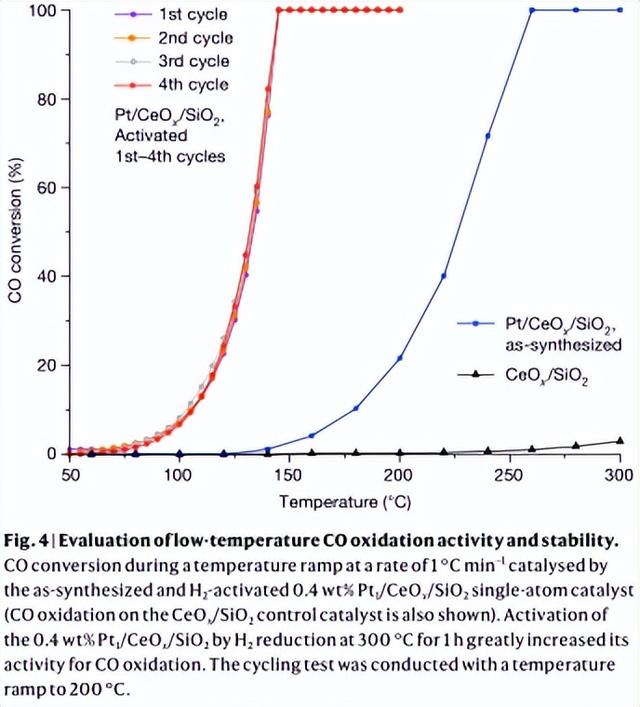
图4
这些观测结果说明了CeOx纳米胶设计策略的价值,该策略通过可扩展的强静电吸附过程实现,将平均尺寸为2 nm或更小的CeOx (x ≈ 1.86)纳米胶岛分散到稳定的高表面积SiO2载体上,然后选择性地将Pt原子定位在这些岛上。CeOx纳米胶岛含有丰富的Ce3 ,在O2或H2环境下,即使在高温下,也能牢固地固定Pt原子和小团簇。然而,实际应用的挑战仍然存在,例如Pt1原子的部分和可逆氧化,以及在氧化条件下较高温度(例如300 oC)下催化剂活性的相关降低。但是,作者的策略是使用功能纳米胶限制金属原子,适用于除Pt (包括Pd和Rh)以外的金属,并且原则上能够产生广泛的单原子和簇催化剂,作者预计它将被证明对许多催化转化有用。












 智钛公众号
智钛公众号 智钛小程序
智钛小程序